Terapia genica – Introduzione alla tecnologia

Con “Terapia Genica” si identifica un trattamento medico basato sul trasferimento di materiale genetico esogeno (sia esso DNA o RNA) direttamente inserito nel nucleo delle cellule di un paziente per prevenire o curare una patologia.

L’obiettivo è quindi gestire la patologia direttamente alle sue basi genetiche fornendo un gene corretto all’organismo che ne ha bisogno.
Storicamente l’idea di utilizzare i geni per prevenire o curare patologie nasce e si sviluppa con il progredire delle tecniche di biologia molecolare e ricombinazione del DNA, esse permettono di intervenire a livello genetico all’interno delle cellule umane eliminando o inserendo geni.
Le prime basi teoriche sono nate intorno agli inizi degli anni 70 pur sottolineando la poca conoscenza dei meccanismi e i rischi di questo approccio in riferimento alla tecnologia e alle scoperte disponibili in quella decade.
Negli anni 80 il lavoro sui virus (gli adenovirus in particolare), che sono poi diventati i vettori per alcune di queste terapie, e le nuove tecniche del DNA ricombinante, con la quale si può costruire pezzi di DNA (Plasmidi) contenenti le sequenze genetiche desiderate, portò alla sperimentazione su una bambina di 4 anni affetta da ADA-SCI (deficit di adenosina deaminasi) della prima terapia genica al mondo (1990). La sperimentazione fu eseguita su una bambina affetta da questa patologia, Ashanti De Silva, all’epoca aveva quattro anni. Il gene difettoso delle cellule del sangue del paziente fu sostituito dalla variante corretta. Il sistema immunitario di Ashanti venne parzialmente ripristinato dalla terapia. La bambina ricevette la sua prima infusione di cellule il 14 settembre 1990, senza complicazioni.
La produzione dell’enzima mancante fu temporaneamente stimolata, ma non furono generate le nuove cellule con geni funzionali. I trattamenti furono ripetuti per dieci volte nei successivi due anni fino al completo ripristino del sistema immunitario della piccola. Oggi Ashanti vive una vita normale. L’Istituto San Raffaele-Telethon per la terapia genica (SR-Tiget) presso l’Ospedale San Raffaele a Milano, diretto dal Prof. Claudio Bordignon è un importante punto di riferimento per le terapie geniche in Italia ed in tutto il mondo. Da questo centro sono state prodotte due terapie geniche approvate dalla Commissione Europea e quindi commercializzate nel territorio europeo. Detengono il record della prima terapia genica approvata al mondo, si tratta di un trattamento contro la Leucodistrofia Metacromatica; questa malattia metabolica è causata da mutazioni di un gene arilsulfatasi-A (ARSA), che portano all’accumulo di particolari sostanze chiamate solfatidi nel cervello e in altre parti dell’organismo, tra cui fegato, cistifellea, reni, milza. Nelle sue forme più gravi, che sono anche le più comuni, questi bambini perdono rapidamente la capacità di camminare, parlare e interagire con il mondo circostante: la maggior parte di loro muore in età infantile e ha a disposizione soltanto cure palliative. Questa malattia colpisce un bambino ogni centomila. La terapia genica Libmeldy viene somministrata con unica infusione, Il trattamento prevede il prelievo delle cellule staminali ematopoietiche del bambino, ovvero quelle staminali che danno origine a tutti gli elementi del sangue. Le cellule vengono poi corrette con un vettore lentivirale modificato che consente di inserire nel loro patrimonio genetico più copie funzionanti del gene ARSA. Queste cellule geneticamente modificate vengono poi reinfuse nel paziente (approccio ex-vivo): la loro capacità di attraversare la barriera emato-encefalica, raggiungere il cervello ed esprimere livelli di enzima funzionante superiori al normale permette di correggere la disfunzione all’origine della malattia in modo continuo e a seguito di un singolo trattamento. L’enzima prodotto in sovrannumero dalle cellule ingegnerizzate viene infatti assorbito e utilizzato anche dalle altre cellule cerebrali che contengono ancora la mutazione nel gene ARSA e che quindi non sono in grado di produrlo da sé. Molto recente è l’approvazione di Casgevy, trattamento per anemia falciforme e talassemia. Prevede l’estrazione di cellule staminali emapoietiche del paziente, vengono modificate geneticamente tramite CRISPR e reinfuse. Dato che nel caso delle due emoglobinopatie in questione (TDT e SCD) la forma adulta di emoglobina non funziona correttamente, la riattivazione della versione fetale va a compensare la carenza e permette di migliorare la gestione della malattia e la sua evoluzione clinica. I dati confermano, infatti, la riduzione o l’eliminazione delle crisi vaso-occlusive nei pazienti con anemia falciforme severa e la riduzione delle trasfusioni nei casi di TDT.
Ulteriori studi sono stati fatti su sindromi genetiche come l’amaurosi congenita di Leber o la sindrome di Wiskott-Aldrich fino all’approvazione della terapia per il trattamento di una rara malattia del fegato (2012). Sono allo studio attualmente diverse decine di terapie geniche tra le quali; Novartis Gene Therapies ha annunciato nuovi datiad interim dello studio clinico di Fase III STR1VE-UE, attualmente in corso con onasemnogene abeparvovec (Zolgensma), i quali hanno dimostrato che i pazienti con atrofia muscolare spinale di tipo 1 (SMA 1) hanno continuato a sperimentare significativi benefici terapeutici, tra cui una sopravvivenza libera da eventi, un miglioramento rapido e sostenuto della funzione motoria e il raggiungimento di specifici traguardi motori, anche nel caso di alcuni pazienti con malattia al basale più aggressiva rispetto agli studi precedenti. AskBio, l’unità di terapia genica di Bayer, sta sperimentando una terapia genica basata su un virus adeno-associato (AAV) per ripristinare la funzione cardiaca. I risultati del trial di Fase I sono stati presentati al Congresso Annuale dell’American Heart Association, e ora è in avvio lo studio di Fase II che avrà una durata di tre anni. Ed ancora restituire la vista a adolescenti che l’hanno persa a causa di una malattia genetica.Biotech italiana AAVantgarde Bio, nata come spin-off della Fondazione Telethon, che entro metà 2024 conta di far partire lo studio clinico sul più avanzato dei due programmi in pipeline: la terapia genica per la sindrome di Usher 1/b, caratterizzata dalla combinazione di cecità e sordità.
Aziende biofarmaceutiche come Moderna, Biontech, Pfizer e decine di altre investono miliardi di dollari nella ricerca di terapie geniche per malattie che oggi non si possono curare o per migliorare le terapie disponibili basate su piccole molecole, pensiamo a tutto il mondo delle neoplasie e alle possibili soluzioni legate alla manipolazione dei geni. Ci sono già sperimentazioni legate alla “vaccinazione” per i melanomi, uno dei tumori della cute più diffusi tra la popolazione.
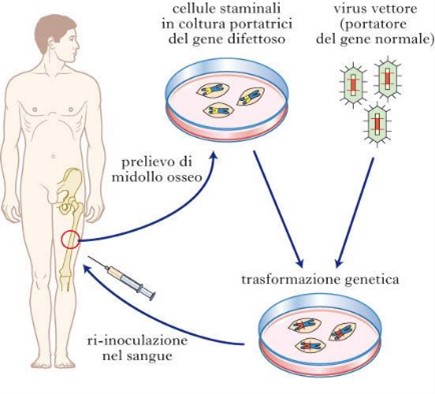
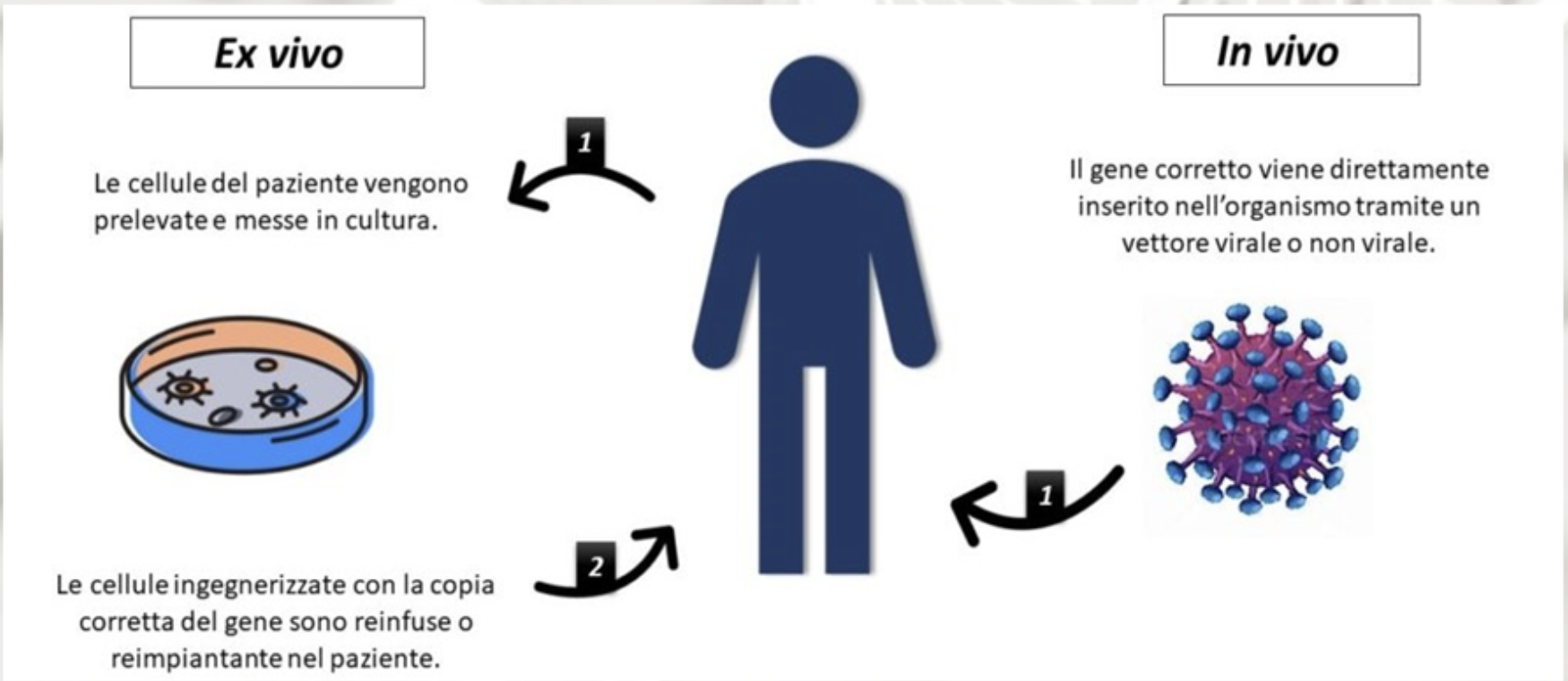
Esistono due principali modalità di somministrazione; in vivo o ex vivo. Nel primo caso il gene viene somministrato direttamente al paziente mediante iniezione o per via sistemica a seconda del bersaglio. Nel caso della ex vivo la correzione dei geni viene fatta all’esterno dell’organismo del paziente. Vengono prelevate delle cellule, modificate e corrette geneticamente e poi reintrodotte nel paziente. Queste tipologie di terapie rientrano nella medicina innovativa moderna, diversi trattamenti sono oggi disponibili presso le strutture ospedaliere italiane pienamente validati ed approvati sia da AIFA che da EMA. Queste terapie sono utilizzate in clinica per trattare alcune malattie rare causate da problemi in specifici geni, sono allo studio, e alcune in fase molto avanzata, terapie geniche per la prevenzione o il trattamento di numerose patologie; dai tumori alle malattie infettive, dal diabete ad alcune malattie cardiovascolari.
CRISPR/Cas9 – pietra miliare
Un deciso passo avanti sulla strada delle terapie geniche è stato fatto con l’introduzione della tecnica CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats) che ha molto semplificato l’editing genetico, ovvero quel tipo di ingegneria genetica in cui parti di DNA vengono inserite, cancellate, modificate o rimpiazzate dal genoma dell’organismo ospite. Questo editing lavora su siti specifici ed è in genere molto preciso nella sua azione. Chi ha inventato e sviluppato questo genere di editing, Jennifer Doudna ed Emmanuelle Charpentier, ha vinto il premio Nobel per la chimica nel 2020.
Ma come funziona questa tecnica? In pratica si utilizza una proteina (Cas9) che è in grado di tagliare un DNA bersaglio in siti specifici e che può quindi essere programmata per modificare in modo preciso il genoma di una cellula animale e/o vegetale.
Una volta che la proteina ha fatto il suo lavoro sul filamento di DNA è possibile eliminare sequenze difettose e sostituirle, se necessario, con sequenze corrette per risolvere patologie genetiche. Si utilizza un RNA guida che identifica il bersaglio e segnala alla proteina Cas9 dove agire. Questo RNA è modificabile in laboratorio per selezionare il bersaglio desiderato.
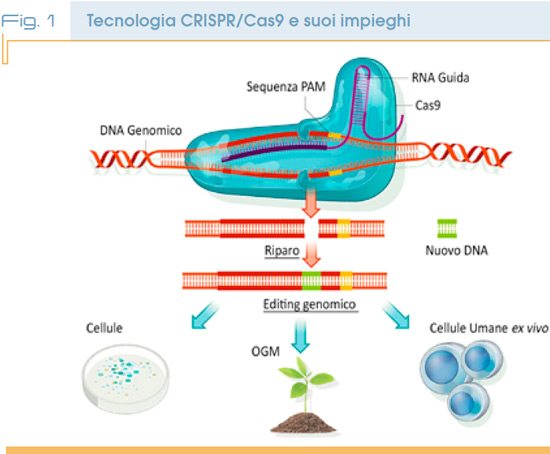
L’identificazione del sistema crisper avvenne nei batteri, qui la proteina Cas9 aiuta la difesa di questi microrganismi dai virus, una sorta di sistema immunitario di prima istanza. Negli anni tra il 2012 e 2013 due gruppi di ricerca statunitensi (Università di Berkeley e MIT Boston) dimostrarono che questo sistema può essere preso in prestito dai batteri ed utilizzato in cellule più evolute come quelle animali e vegetali.
Questa tecnologia ha aperto una serie di studi in campo biomedico poiché per la prima volta si è riusciti ad introdurre modificazioni desiderate nel genoma in modo semplice, efficace, veloce ed economico. Lo dimostra in numero crescenti di laboratori in tutto il mondo che utilizza crisper sia per la ricerca di base che per scopi applicativi, la sua robustezza la sta rapidamente spingendo verso sperimentazioni cliniche sempre più innovative verso malattie genetiche considerate incurabili. A riportarci con i piedi per terra ci sono gli effetti collaterali legati a questa tecnologia, in particolare i cosiddetti errori di taglio (detti off-target) che avvengono in parti del genoma che non è considerato bersaglio della terapia. Per correggere questi errori sono allo studio diverse soluzioni legate spesso all’utilizzo di RNA più performanti e precisi.
Per utilizzare una terapia genica abbiamo bisogno di un vettore, spesso questo è proprio un virus che ci permette di far arrivare a destinazione il materiale genetico che abbiamo creato. Sono utilizzati diversi tipi di virus, adenovirus (vaccini in generale), lentivirus o retrovirus per CRISPR. Questi arrivano alla cellula bersaglio e rilasciano il contenuto di materiale genetico (sia esso DNA o RNA) che andrà a svolgere la sua funzione.
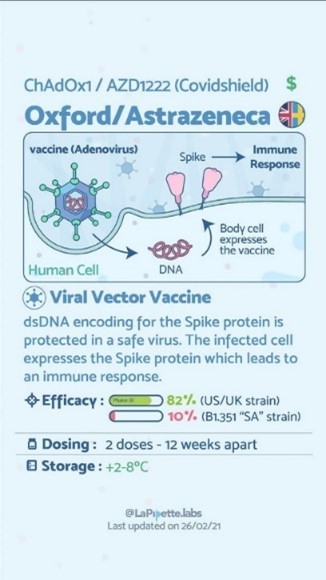
Un esempio di questa tecnologia è il vaccino Astrazeneca AZD1222 (nome commerciale Vaxzevria) che utilizza un adenovirus ChAdOx1 - Chimpanzee Adenovirus Oxford1 modificato per impedirne la replicazione, con cui è possibile far arrivare del dsDNA (double stranded DNA, doppia elica del DNA) all’interno della cellula. Una volta all’interno partono i meccanismi di trascrizione del dsDNA in una proteina (Spike del SARS-Cov-2) che viene riconosciuta come antigene dal nostro sistema immunitario, il quale crea anticorpi contro questa proteina. Quando l’organismo dovesse venire a contatto con il virus la cui proteina è contenuta, lo riconoscerà e svilupperà anticorpi per affrontarlo. La glicoproteina Spike è ovviamente sulla superficie del virus SARS-Cov-2 ed è la prima che viene a contatto con le cellule ed i recettori del sistema immunitario umano, questo permette una risposta immunologica normale.
Astrazeneca ha recentemente ritirato dal commercio questo vaccino, ritenuto ormai obsoleto rispetto alle varianti del SARS-Cov-2 presenti oggi. Il vaccino era infatti basato sul primo virus individuato che attualmente non circola più, soppiantato da varianti più efficaci.
Si è poi sperimentato l’uso dei Lentivirus, che spesso sono fortemente patogeni (l’HIV è veicolata da un Lentivirus). La loro capacità di passare “inosservati” dal nostro sistema immunitario li rende dei fantastici vettori virali. Essendo patogeni la loro gestione è un po' più complessa rispetto agli adenovirus ma sono molto promettenti per veicolare terapie geniche. La terapia genica messa a punto dall’SR-Tiget si basa su un vettore virale lentivirus responsabile dell’HIV modificato per impedirne la replicazione ma mantenendo intatte le sue capacità di mimetismo al sistema immunitario e la sua lenta replicazione.
Le LNP (Lipid Nano Particle) rappresentano una classe di vettori che vide la luce all’inizio degli anni’80. I liposomi, sfere lipidiche cave spesso costituite da due o tre tipi di lipidi, furono identificati come portatori di farmaci antitumorali dallo scienziato esperto in nanoparticelle Pieter Cullis (University of British Columbia): i farmaci potevano penetrare in questi liposomi e rimanere intrappolati nel loro nucleo cavo. Una volta iniettati in animali malati di tumore, i liposomi possono viaggiare attraverso il sistema vascolare, penetrare nella zona interessata dal tumore, entrare nelle cellule e liberare il farmaco. Il loro progresso e utilizzo è stato rallentato da problemi di stabilità e produzione. Il primo farmaco a base di liposomi fu approvato dalla FDA statunitense nel 1995, ma a quel punto Cullis e molti nel campo erano passati a una nuova sfida: usare le LNP per trasportare acidi nucleici come DNA e RNA. Per fornire terapie con acidi DNA o RNA serviva bilanciare gli acidi nucleici caricati negativamente, erano quindi necessari lipidi caricati positivamente, ma non ci sono lipidi cationici in natura e i lipidi caricati positivamente sono citotossici, causano la distruzione delle membrane cellulari. Durante la fine degli anni ’90 e durante il primo decennio degli anni 2000, una soluzione è stata individuata in nuovi lipidi carichi positivamente solo a determinate condizioni. Cullis, i suoi colleghi di Inex Pharmaceuticals e lo spin-off Inex Protiva Biotherapeutics svilupparono lipidi ionizzabili che sono carichi positivamente a un pH acido, ma neutri nel sangue. Il gruppo creò anche un nuovo modo per produrre nanoparticelle con questi lipidi, utilizzando la microfluidica. Le nanoparticelle lipidiche, a differenza dei liposomi cavi, contenevano lipidi e acidi nucleici impaccati in modo molto fitto. Il processo era semplice in teoria, ma le prime versioni di lipidi ionizzabili erano ancora tossiche e le prime formulazioni delle nanoparticelle tendevano ad accumularsi in alcuni organi dopo ripetute iniezioni. Nell’ultimo decennio, lo studio di terapie basate sull’mRNA ha raccolto miliardi di dollari di investimenti portando alla creazione di nanoparticelle stabili, non citotossiche e facilmente producibili sotto GMP. Per il vaccino Pfizer/Biontech sono state utilizzate della LNP da 100 nanometri.La capacità di produrre questi vettori in ambiente GMP (buona pratica di fabbricazione) le ha rese una buona alternativa ai vettori virali verso i quali presentano meno problematiche immunologiche e patologiche. Non sono prive di problemi, sono infatti poco stabili (il vaccino Pfizer/Biontech va conservato a temperature molto basse proprio per questo motivo) e non permettono il rilascio preciso del farmaco nelle cellule d’interesse. Il mondo dei vettori è in continua evoluzione proprio perché sono il punto più complesso per queste terapie. Come si può vedere dalla tabella qui a fianco la scelta di vettori non virali è varia e dipende dall’organo bersaglio e dalla tipologia di informazione utilizzata (RNA o DNA), dal costo del vettore e dalla facilità di fabbricazione sotto regole GMP.
CAR-T: una terapia genica contro i tumori
Le terapie Car-T (Chimeric Antigen Receptor T cell therapies), sono un innovativo strumento per combattere alcune forme di neoplasie, il concetto base della terapia è semplice; istruire i linfociti T, le cellule adibite alla produzione di immunoglobuline (anticorpi) o tossine in grado di indurre la distruzione delle cellule batteriche o di quelle cellule riconosciute come estranee all’organismo, od altre cellule specializzate del sistema immunitario ad attaccare in maniera mirata e specifica le cellule tumorali. Questa idea risale ad oltre venti anni fa quando diversi ricercatori iniziarono ad osservare un’ottima attività negli esperimenti in vitro.
L’FDA, capite le potenzialità di questa tecnica, creò una corsia preferenziale che permise di arrivare in tempi relativamente brevi all’utilizzo clinico.
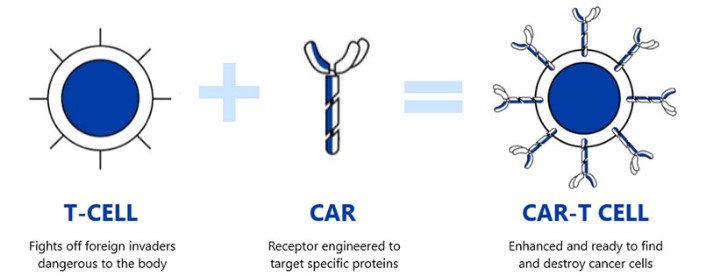
I linfociti T modificati, in grado di produrre l’anticorpo CAR (da qui deriva il termine Car-T), saranno in grado di riconoscere più efficacemente le cellule tumorali bersaglio per poi distruggerle. Possiamo quindi considerare questi linfociti T delle truppe speciali, addestrate a riconoscere e distruggere quel nemico che prima non riuscivano a vedere.
Rispetto alla chemioterapia, che è indiscriminata nel distruggere le cellule eliminando anche quelle sane che servono, è molto più specifica ed attacca solo le cellule tumorali, riducendo gli effetti collaterali e incrementando l’efficacia terapeutica.
Sono terapie molto efficaci ma che hanno delle difficoltà soprattutto nella loro produzione, ad oggi sono disponibili solo in forma autologa ovvero partendo dalle cellule del paziente con un processo di produzione personalizzato complesso.
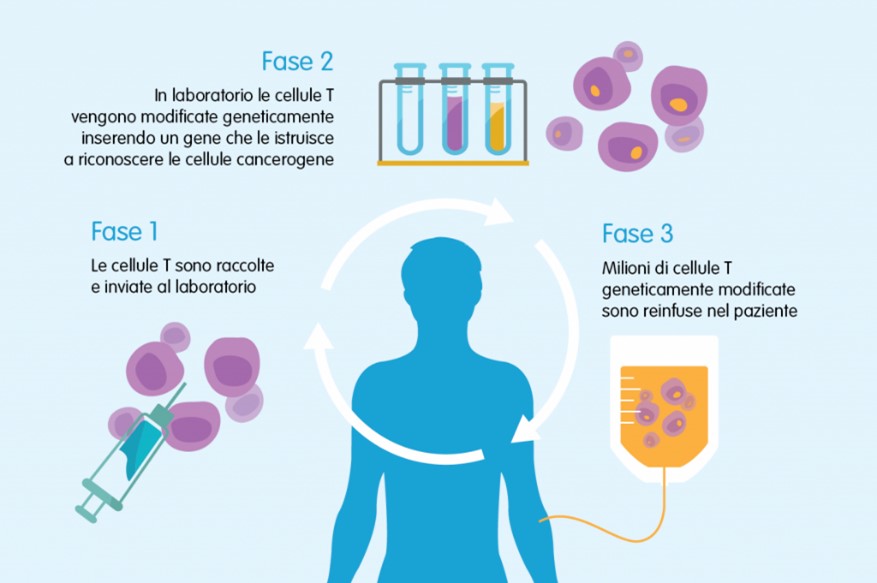
La terapia fa quindi perno sul paziente a cui devono essere estratti i linfociti T (leucoaferesi), poi modificati tramite ingegneria genetica con l’ausilio di un vettore virale che inserisce l’informazione genetica per la formazione del recettore CAR. Questa modifica è permanente, basta quindi “coltivare” i globuli bianchi fino al raggiungimento della quantità necessaria per il trattamento. Tutto il processo è costellato di controlli e verifiche per garantire il prodotto finale. Il tutto dura un paio di settimane, la terapia è applicata in un’unica soluzione per infusione. Come tutti i farmaci anche le Car-T sono sottoposte ai processi di valutazione per avere la finale validazione ed approvazione ad essere utilizzate sul mercato.
Anche in questo caso sono possibili effetti collaterali, riscontrabili su circa il 25% dei pazienti. La sindrome da rilascio delle citochine è uno dei problemi più comuni, queste molecole veicolano la risposta infiammatoria del nostro organismo, ecco spiegato perché queste procedure sono somministrate in ospedali con terapia intensiva attrezzata per fronteggiare qualsiasi situazione si possa presentare.
Questo effetto collaterale e la sua risoluzione mette in luce, come del resto ha fatto il Covid19, la relativa scarsità di posti in terapia intensiva negli ospedali italiani.
Il secondo problema che può insorgere dopo l’infusione è quello della tossicità neurologica; cefalea, mal di testa, disorientamento ed agitazione, afasia e crisi epilettiche sono state osservate, in alcuni casi si è arrivati ad encefalopatia grave. Tutte problematiche risolvibili con l’utilizzo di corticosteroidi e/o mAb (anticorpi monoclonali) atti ad inibire le citochine probabili responsabili di questi fenomeni. Nell'Unione Europea (UE) sono stati approvati sei prodotti a base di cellule CAR-T. Questi farmaci sono utilizzati per il trattamento di tumori del sangue come la leucemia a cellule B (LLA), il linfoma a cellule B, il linfoma follicolare, il mieloma multiplo e il linfoma a cellule mantellari in pazienti il cui tumore si è ripresentato (recidiva) o ha smesso di rispondere al trattamento precedente.
Rischi Terapie geniche
Come tutte le procedure mediche anche queste terapie possono avere delle controindicazioni e dei rischi potenziali. Nel 2021 la morte di un paziente affetto da Miopatia miotubolare che stava partecipando ad uno studio clinico per testare la sicurezza e l’efficacia della terapia genica sperimentale AT132, riaprì il dibattito sulla sicurezza di queste terapie. Si trattò purtroppo del quarto decesso tra i pazienti arruolati nel trial clinico. I primi tre erano avvenuti tra marzo e agosto del 2020 portando l’agenzia regolatoria statunitense (FDA) a sospendere la sperimentazione clinica. Lo studio era poi ripartito a dicembre dello stesso anno con una modifica della dose di trattamento, ridotta della metà. Per nessuno dei quattro giovani pazienti è stato chiarito il legame tra la loro morte e il trattamento con la terapia genica. Tutti i partecipanti deceduti hanno presentato danni epatici in seguito alla somministrazione della terapia a dose più elevata, questi sono evoluti in una insufficienza epatica. La causa della morte in due dei partecipanti è stata la sepsi e nel terzo un’emorragia gastrointestinale. FDA ha quindi convocato un comitato di esperti per discutere sulla sicurezza delle terapie geniche e costruire dei protocolli che permettano di ridurre al minimo i rischi. Sono diversi i problemi che si possono riscontare, non è possibile escludere del tutto reazioni anomale del sistema immunitario, questo potrebbe riconoscere come antigene il vettore virale utilizzato e reagire. Questo è un problema legato anche alle vaccinazioni, se l’adenovirus utilizzato come vettore virale venisse riconosciuto come estraneo e si sviluppassero anticorpi, la sua efficacia verrebbe compromessa. Un secondo problema è legato al bersaglio, vi è la possibilità di colpire una cellula sana o una porzione di DNA non corretta. Vi sono poi delle questioni etiche legate all’utilizzo di terapie geniche dette germinali, effettuale sulle cellule della linea germinale e che introduce quindi delle modifiche che verranno trasmesse alle successive generazioni. Potremmo allargare questo concetto e chiederci come sarà possibile distinguere un uso etico da uno non etico.
Per esempio, introducendo terapie che modifichino caratteristiche fisiche dando vantaggi o modificando in generazioni future caratteri che queste non hanno deciso. Si tratta di discussioni sul filo del rasoio, spesso lontane o molto fumose ma restano nodi da sciogliere. Al momento questi nodi sono verificati dall’ISS (Istituto Superiore Sanità) che attiva uno specifico laboratorio (OMLC) che afferisce a livello europeo al Direttorato per la qualità dei medicinali e delle cure sanitarie (EDQM). il Gene Therapy Working Group, che nasce per migliorare la collaborazione tra i diversi laboratori OMCL europei. Questo gruppo si riunisce annualmente per condividere i risultati ottenuti sulla pianificazione dell’anno precedente e valuta eventuali nuove problematiche presenti. Qualsiasi terapia genica utilizzata in ambito europeo deve passare il vaglio della EMA (European Medicines Agency) e alle organizzazioni nazionali di ogni stato, per l’Italia l’AIFA.
Ti è piaciuto questo articolo? Supporta la nostra associazione: associati oppure effettua una donazione.
Il tuo sostegno è per noi importante!
Energia Blu, analisi e riscoperta del nucleare civile
di Luca Proietti
Il giorno Martedì 11 marzo 2025 si è tenuta in Università Cattolica di Milano la conferenza "Energia…
Gli autocrati ringraziano: l’Occidente e l’arte della complicità
di G. Stassi
L’attuale crisi geopolitica, con la guerra in Ucraina come epicentro, non è un fulmine a ciel…
Quorum o non quorum? Il vero scontro è sulla democrazia
di M. Pulieri
L’8 e 9 giugno 2025 i cittadini italiani saranno chiamati a esprimersi su cinque quesiti referendari…